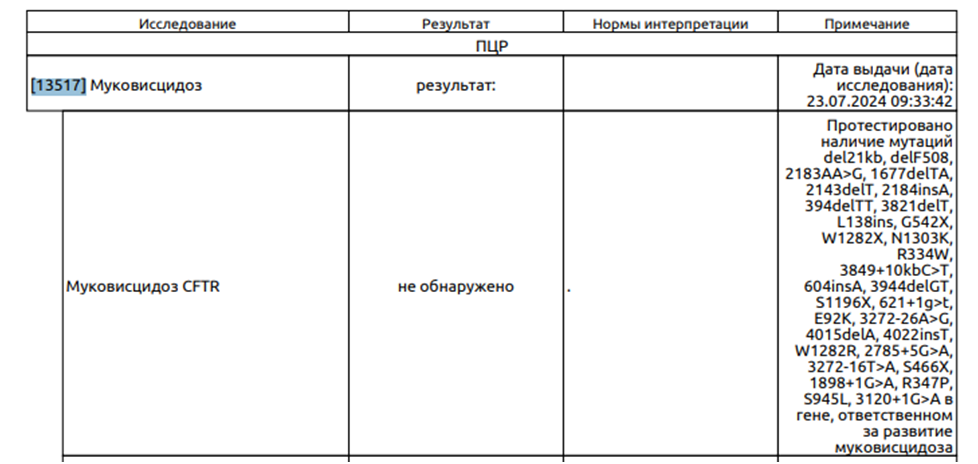
Описание исследования
Комплексное генетическое исследование, которое позволяет выявить 30 наиболее часто встречающихся на территории России мутаций гена CFTR, приводящих к развитию тяжелого наследственного заболевания муковисцидоза.
Муковисцидоз (синоним – кистозный фиброз) – одно из наиболее распространенных аутосомно-рецессивных наследственных заболеваний человека. Он характеризуется нарушением функции эпителия дыхательных путей, кишечника, поджелудочной железы, потовых и половых желез.
Причиной развития муковисцидоза являются мутации в гене CFTR (cysticfibrosistransmembraneregulator), кодирующем АТФ-связывающий белок, который формирует канал для ионов хлора в клеточных стенках. Мутации приводят к нарушению транспорта электролитов и ионов хлора через мембраны эпителиальных клеток, что сопровождается усилением секреции густой слизи и закупоркой выводящих протоков различных желез.
Существует несколько форм муковисцидоза:
- смешанная (поражаются одновременно органы дыхания и пищеварительный тракт);
- бронхолегочная (поражаются преимущественно органы дыхания);
- кишечная (поражается преимущественно желудочно-кишечный тракт);
- мекониевая непроходимость кишечника;
- атипичные формы, связанные с изолированными поражениями отдельных желез внешней секреции.
В настоящее время в РФ диагноз "муковисцидоз" ставится одному из 9 000 новорождённых (для сравнения: в Европе муковисцидоз диагностируется с частотой 1 : 2 000 – 3 000 новорождённых). Однако принятая в России форма массового скрининга новорождённых несовершенна и иногда не позволяет выявить заболевание на доклинической стадии.
В каждой клетке нашего организма имеется две копии гена CFTR. Одна копия достается от отца, а вторая от матери. Заболевание муковисцидоз аутосомно-рецессивное, т. е. развивается оно только при условии, что ребенок получает и от отца, и от матери мутантный ген CFTR. При этом родители, у которых вторые копии гена CFTR нормальные, не страдают муковисцидозом и порой даже не догадываются о его носительстве. По статистике, в европейской популяции носителем мутаций гена CFTR в среднем является каждый 25-й человек.
Насчитывается примерно одна тысяча различных мутаций в гене CFTR. Они встречаются с различной частотой в разных популяциях. Некоторые нарушения в гене могут не иметь никаких проявлений. Но большая часть мутаций вызывает патологический эффект, т. к. приводит к нарушению функционирования белка.
В данное комплексное исследование включен анализ гена CFTR на 30 мутаций, наиболее распространенных на территории РФ, Восточной Европы и Скандинавии и связанных с развитием тяжелых клинических форм муковисцидоза. Исследование позволяет выявлять до 95 % всех возможных больных, что существенно превышает разрешающие способности утвержденного в России скрининга.
Исследование поможет не только подтвердить или опровергнуть диагноз "муковисцидоз", но и выявить носительство мутации у здоровых людей. Особенно важно проводить генетическое тестирование в семьях, в которых есть больные муковисцидозом, поскольку у пары, где оба родителя являются носителями мутаций, вероятность рождения больного ребенка составляет 25 %.
До сих пор муковисцидоз считается неизлечимым заболеванием, но ранняя диагностика и адекватная терапия значительно улучшают прогноз заболевания и продляют пациенту жизнь.
Подготовка к исследованию
Специальной подготовки не требуется.
Интерпретация исследования
В ходе анализа проводится исследование 30 значимых генетических маркеров гена CFTR, что позволяет обнаружить наиболее распространенные мутации, приводящие к развитию заболевания.
N (норма) / N (норма) – мутации не обнаружены.
N / M (мутации) – выявлена гетерозиготная мутация, скрытое носительство.
M (мутация) / M (мутация) – выявлена гомозиготная мутация, подтверждение диагноза "муковисцидоз".
По итогам комплексного исследования выдается заключение врача-генетика с интерпретацией результатов.
Муковисцидоз
(CYSTIC FIBROSIS; CF; OMIM 219700)
Муковисцидоз – наследственное аутосомно-рецессивное заболевание, характеризующееся
поражением желез внешней секреции, тяжёлыми нарушениями функций органов дыхания и
желудочно-кишечного тракта.
В ходе исследования в гене CFTR протестированы следующие 30ть мутаций:
• c.54-5940_273+10250del21kb (CFTRdele2,3), c.1521_1523delCTT (F508del),
c.2051_2052delAAinsG (2183AA>G), c.1545_1546delTA (1677delTA), c.2012delT
(2143delT), c.2052dupA (2184insA), c.262_263delTT (394delTT), c.3691delT (3821delT),
c.413_415dupTAC (L138ins), c.1624G>T (G542X), c.3846G>A (W1282X), c.3909C>G (N1303K), c.1000C>T
(R334W), c.37182477C>T (c.3717+12191C>T; 3849+10kbС>Т), c.472_473insA (604insA),
c.3816_3817delGT (3944delGT), c.3587C>G (S1196X), c.489+1G>T (621+1G>T), c.274G>A(E92K), c.3140-
26A>G (3272-26A>G), c.3883delA (4015delA), c.3891dup (4022insT), c.3844T>C(W1282R), c.2657+5G>A
(2789+5G>A), c.3140-16T>A (3272-16T>A), c.1397C>G (S466X), c.1766+1G>A (1898+1G>A),
c.2988+1G>A (3120+1G>A), c.1040G>C (R347P), c.2834C>T(S945L).
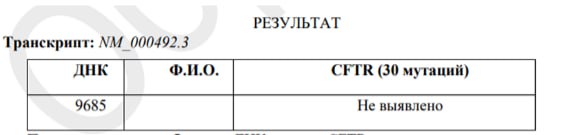
При исследовании образцов ДНК в гене CFTR частых патогенных вариантов, птветственных за развитие заболевания, не обнаружено.
! Однако важно понимать, что отсутствие наиболее частых патогенных вариантов в гене CFTR не исключает вероятность носительства редких.
Возможен анализ всей кодирующей последовательности гена CFTR.
При необходимости получения дополнительной информации рекомендуется очная консультация врача-генетика.